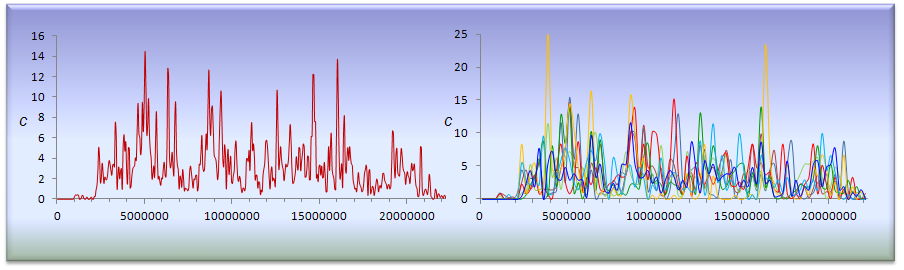
The many landscapes of recombination in Drosophila melanogaster (2012) PLoS Genet. 8(10):e1002905
High-Resolution Recombination Maps in Drosophila melanogaster
Until recently, there were at least three limitations associated with most recombination maps. First, population genomic analyses can now analyze patterns of selection at the scale of single genes, or even focus on specific gene regions, but most whole-genome genetic maps localize recombination events with much less detail. Second, most species' maps based on direct measures of recombination are obtained either from two specific, usually highly diverged, strains, or compiled from crosses in different laboratory/natural conditions that can also influence recombination rates. These genetic maps are customarily assumed to represent a monomorphic description of recombination for a given species, even when there is ample evidence of intra-specific natural variation. Third, the process of meiotic recombination associated with the repair of double strand breaks (DSB) has two possible outcomes with diverse evolutionary consequences: crossing over (CO) and non-crossing over (NCO). Unlike CO, NCO results in the exchange of only small tracts of a chromosome, interrupting linkage disequilibrium in a much localized manner while having no effect on linkage disequilibrium at longer intervals, thus NCO plays a stronger role than CO at short physical distances. In multicellular organisms, CO maps are used as proxy for total recombination, and the consequences of NCO are often overlooked, despite its potential influence on total recombination.
Our lab is generating high-resolution, whole-genome maps of crossing-over (CO) and non-crossing over (NCO) events in D. melanogaster using large-scale, next-generation Illumina sequencing approaches. Recently, we published a recombination map in D. melanogaster by genotyping a total of 139 million informative SNPs, with 32,511 CO events mapped at a resolution down to 2 kb. (PLoS Genetics, 2012).
This study represented the first integrated, high-resolution description of genomic and population variation in recombination, which also distinguished between the two outcomes of meiotic recombination. Our results and conclusions will help to characterize the molecular basis of the observed variation in recombination across genomes and have an immediate impact on population genetic analyses of selection, laying the foundation for a new generation of population models that will better capture natural variation in recombination and its consequences.
Comeron JM, Ratnappan R, Bailin S. 2012. The many landscapes of recombination in Drosophila melanogaster. PLoS Genetics 8(10):e1002905
We are expanding our initial study to investigate variation in recombination under different biotic and abiotic conditions (age, temperature, etc.).
Molecular Causes of Recombination Variation
The lab is investigating the genetic, genomic and environmental factors associated with recombination variation across the genome and among individuals of the same species. We study D. melanogaster and combine classical genetics, molecular biology, machine-learning and next-gen sequencing techniques. This research is currently funded (-2019) by NSF (DEB1354921).
Adrian A, Cruz Corchado J, Comeron JM. 2016. Predictive models of recombination rate variation across the Drosophila melanogaster genome. Genome Biol. Evol. 8(8): 2597-2612
Adrian A, Comeron JM. 2013. The Drosophila early ovarian transcriptome provides insight to the molecular causes of recombination rate variation across genomes. BMC Genomics 14: 794
More information coming soon... [see Publications]
Evolutionary Consequences of Recombination Variation across and among Genomes
We are investigating how variation in recombination rates influence patterns of selection and polymorphism using population genetics theory, models and simulations. We pay particular attention to the consequences associated with the unavoidable input of deleterious mutations and removal by natural selection (a process called Background Selection, BGS). These studies provide insight into the relative contribution of mutation, selection and drift to the observed patterns of nucleotide variation across Drosophila genomes.
Comeron JM. 2017. Background selection as null hypothesis in population genomics: Insights and challenges from Drosophila studies (in review)
Comeron JM. 2014. Background selection as baseline for nucleotide variation across the Drosophila genome. PLoS Genetics 10(6): e1004434.
More information coming soon... [see Publications]
Funding Sources
NSF
NSF_DEB1354921
NSF_DEB0344209
NIH
NIH_RC2GM092501

Roy J. Carver Charitable Trust
C05-2258